
A Practical Guide to Dietary Supplement Labelling Requirements
Introduction: The FDA offers an online dietary supplement labeling guide that provides labeling requirements for supplement manufacturers. The guide covers issues such as Supplement Facts Panel (SFP) labeling, ingredient labeling, and health claims. In the USA, there are strict requirements for supplement labels. The FDA (Food and Drug Administration) requires all supplement labels to list
... Read moreSteps to Achieve GFSI Certification for Your Business

https://youtu.be/XRZ87ifRki0 Ensuring the safety and quality of your food products is crucial for building trust with your customers and expanding your business. Achieving Global Food Safety Initiative (GFSI) certification can help you meet international food safety standards and demonstrate your commitment to delivering safe products. Moreover, GFSI certification offers several benefits. These include increased consumer
... Read moreFDA Prior Notice for Food Importers: Unlocking the FDA Guidelines

Introduction Prior notice is a requirement for all food manufactured, processed, packed, or held outside the United States that is imported into the United States. It ensures that the U.S. Food and Drug Administration (FDA) has the opportunity to evaluate the safety of a food shipment before it enters U.S. commerce and provides an opportunity
... Read moreGRAS, NDI, ODI Food Additives & Generally Recognized as Safe GRAS

Introduction Food additives are chemicals added to food products to enhance flavor, texture, or color. Fortunately, there is a list of food additives that the Food and Drug Administration (FDA) has determined to be “generally recognized as safe” (GRAS). In this guide, you’ll find an updated and comprehensive list of GRAS food additives. Food classification
... Read moreFDA Food Safety Requirements FSMA Traceability BRC Standards

Introduction: Meeting FSMA compliance requirements is one of the most important things a manufacturer can do to protect their business. The Food Safety Modernization Act (FSMA) was passed by Congress in 2011, and it places new regulations on food producers and manufacturers. While there are many ways to meet FSMA regulations, one way is through
... Read moreVHP Consulting 5 things about the VHP Notification Program

Introduction Health Canada oversees the importation and sale of Veterinary health products (VHPs) in Canada through the VHP Notification Program. This program replaces the previous voluntary Interim Notification Pilot Program (INPP). The VHP Notification Program is a tool that allows manufacturers to notify Health Canada about the sale of their VHPs and changes in their
... Read moreExplore Health Canada’s Medical Device Regulations (MDL, MDEL & SaMD)
Introduction: Medical devices are an important part of healthcare and play a critical role in the treatment of patients. Medical devices are used to diagnose, monitor, or treat medical conditions in Canada and around the world. The Canadian Medical Devices Regulations provide a framework for regulating medical devices that meet Health Canada’s standards. The regulations
... Read moreHACCP Implementation: A Practical Guide for Food Businesses

HACCP, which stands for Hazard Analysis and Critical Control Points, is the cornerstone of food safety plans in the food industry. It is a concept designed to identify and prevent or eliminate any potential hazards that may put consumers at risk of food-borne illnesses. This guide will take an in-depth look into what HACCP is
... Read more5 FDA Warning Letters: Food Manufacturers of CBD Food Products

Introduction On November 21, 2022, the Food and Drug Administration (FDA) Center for Food Safety and Applied Nutrition issued warning letters to food manufacturers who were selling products containing cannabidiol (CBD). These letters were issued in response to an online search for companies selling foods or beverages containing CBD as a continued FDA CBD enforcement
... Read moreSafe Quality Food Certification SQF Food Safety Program

Food is one of the most important parts of our lives. We eat it to stay alive, and we eat it to enjoy life. Unfortunately, foodborne illnesses are a major issue in Canada, taking a toll on our health and economy. One way to reduce the risk of foodborne illnesses is to certify your food
... Read moreFDA UDI Labeling Requirements for FDA Class 1 Medical Device in 2023

Introduction: FDA regulations for Class 1 medical devices can be daunting and confusing to navigate. However, with this easier-to-understand overview, you’ll gain insight into the prerequisites for device categories, UDI labeling requirements, and more. With the proliferation of medical devices worldwide, it is important to stay on top of applicable UDI labeling requirements. This
... Read moreUnderstanding MDALL/MDEL & How It Transforms Your Business (MDL, MDALL, MDEL Listing)
Navigating medical device regulations set by Health Canada can be challenging. However, knowing the correct classification of your medical device is essential. Proper classification ensures your device complies with regulatory requirements. MDALL provides a strong solution for businesses looking to improve operations. It supports automated workflows, better customer service, and higher productivity. Using MDALL can
... Read moreMedical Device License (MDL MDEL SaMD) Under the Microscope
The Medical Device License is a legal document required to manufacture, sell, and distribute medical devices. The Medical Device Licence (MDL) and the Medical Device Establishment Licence (MDEL) are the licenses issued by Health Canada, the Health Agency that regulates the medical devices sold in the Canadian market. These licences are referred to as “medical
... Read moreCFIA Enforcement of Nutrition Facts Table & CFIA Labeling Requirements
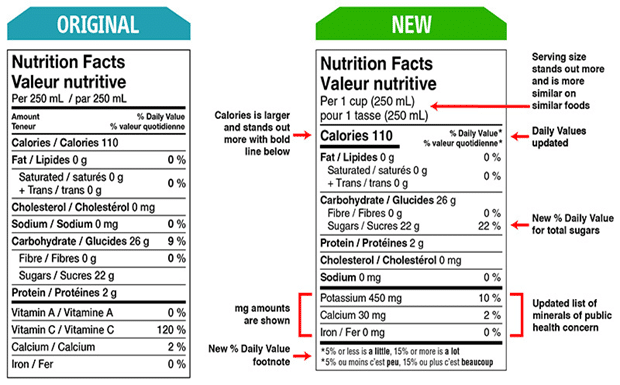
The Canadian Food Inspection Agency (CFIA) protects Canadians’ health and safety by ensuring food that reaches customers is safe, nutritious, and labeled. The CFIA’s responsibilities also include enforcing federal food safety and nutrition regulations. As of July 1, 2016, the CFIA has begun to enforce the new Nutrition Facts Table (NFT) and food labeling regulations. All
... Read moreFSVP Compliance: Regulatory Assessment Record Keeping 2023

U.S.A RRA FDA Draft Guidance: In a recent U.S. Food and Drug Administration (FDA) draft guidance detailing its implementation of Remote Regulatory Assessments (RRAs). Questions and answers outlined the regulatory oversight during and after the COVID-19 pandemic. The draft, “Conducting Remote Regulatory Assessments: Questions and Answers,” outlines the FDA’s use of RRAs to continue
... Read moreHow the Cosmetic Notification Form Works in Canada

Top Canada Cosmetic Regulations Cosmetic Notification Form (CNF) Canada is one of the most popular tourist destinations in the world, and it’s no surprise that the country has some of the most stringent cosmetic regulations in the world. In this blog post, we’ll look at some of the top Canadian cosmetic regulations and what you
... Read moreFood Labelling & Health Canada Packaging Update for 2023

Health Canada Announcement: The Minister of Health has introduced new front-of-package nutrition labeling laws for prepackaged food products, and they will apply to packaged foods high in saturated fat, sugars, and salt. A front-of-package emblem will need to be shown on these prepackaged foods in order to comply with the new requirements. Food products
... Read moreInside Scoop FDA Medical Device User Fees (MDUFA) for 2023
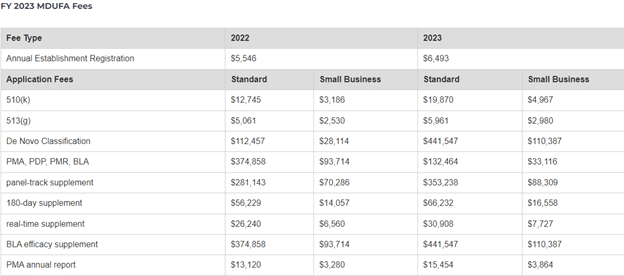
The FDA or U.S. Food and Drug Administration recently announced the Fiscal Year Medical Device User Fee (MDUFA) amendments. The fiscal year will begin on October 1st, 2022, and end on September 30th, 2023. All medical device facilities will have to pay these fees to remain FDA-compliant. In this blog, we’re going to elaborate on
... Read moreGRAS Consultants: NDIN or FDA GRAS Certification?

Say you have a new ingredient and are ready to enter the US food market. To ensure this ingredient is considered approved (and the product will not be considered adulterated upon formulation) for use, you have a decision to make: Do you wish to obtain ingredient approval through GRAS (Generally Recognized As Safe) or via
... Read moreKey Natural Health Product Regulations Canadian Businesses Should Know

The Canadian government has a set of regulations in place for natural health products (NHPs). These regulations help to ensure the safety and quality of NHPs sold in the country. If you’re planning on selling NHPs in Canada, it’s important to be aware of these regulations. Note: The Natural Health Products Directorate (NHPD) has changed
... Read moreInside Scoop MDL VS MDEL & SAMD: Medical Device License Unlocked
The Medical Device License is a legal document that is required for the manufacture, sale, and distribution of medical devices. A medical device license is a type of intellectual property (IP) license that is issued by the FDA to authorize a manufacturer to sell its medical device in the United States. The MDL is
... Read moreDrug Identification Number (DIN) Unlocked Inside Scoop
Many drug industries are working in every country. To sell any OTC drug in Canada is not possible without its legal authorization. A computer-generated eight-digit number is issued to every drug in Canada from Health Canada before it comes to market. This eight-digit number is known as Drug Identification Number (DIN). Drug Identification Number
... Read moreFSVP Plan: Levels of FSMA-FSVP Certification

The FSVP Plan is a requirement for many companies to be FSMA compliant. It is the responsibility of the company to develop and maintain a plan that outlines how it will verify the safety of its food supply chain. The FSVP-FSMA certification is required for all companies that want to export food products to other
... Read moreHow Does FDA Classify Software As A Medical Device (SaMD)?

Nowadays almost all the facets of healthcare are well-equipped with the modern and latest technology tools, including software systems. The use of the software is fully incorporated into digital services for both medical and non-medical uses. One of three forms of software connected to medical devices is software that, on its own, qualifies as
... Read moreHealth Canada’s Regulatory Enrollment Process (REP): Top 4 things to know
Industry people and experts considering working with a company not registered with Health Canada should be aware of the regulatory enrolment process. The REP is a regulatory enrollment process for medicinal products. It is an essential step in the pathway to market approval. A drug company must apply to Health Canada’s Natural and Non-prescription Health
... Read moreWhat are (HACCP & PCP) Requirements for GFSI Certification?

https://youtu.be/XRZ87ifRki0 Since the publishing of the Safe Foods for Canadians Regulations in 2017, there is now a requirement for a Preventative Control Plan (PCP) for your food facility or food program. This blog aims to explain several different programs and certifications that a food facility may want to consider for your sites or products. HACCP,
... Read moreNatural Health Products (NHP/NPN) Panel Top 19 Questions Answered

1. What is the NHP Licensing Process/what is needed for it? This process involves the submission of a product license application to Health Canada. Ø Documents normally contained within a submission are Cover Letter and a Product license Application form. Other documents may be required based on the classification of the product and who is
... Read moreSupplemented Foods: Food & Drug Regulations Unlocked in 2023

Have you been anticipating the Food and Drug Regulations (FDR) adding supplemented foods as an official category of food? As a result of the July 20th amendments, these food products can now be sold in Canada as of July 21st, 2022. This has been the result of over 10 years of contributions which began
... Read moreRegulations amending the Natural Health Products Regulations (NHPR) | What you should know!

What are the latest Natural Health Product Amendments (NHPR) Health Canada will be amending the Natural Health Products Regulations (NHPR) so they can modernize the requirements. These new requirements for the labelling of NHPs aim to improve consistency and legibility. While also making the information clear while being aligned with pre-established rules for comparable non-prescription
... Read moreNutrition Symbol Front-of-Food-Package Label Regulations

Health Canada recently announced that Front-of-package labelling changes for foods are set to come into effect on July 20, 2022, but the industry will have until January 1, 2026, to comply. A new front-of-package nutrition symbol will be required for prepackaged foods high in sodium, sugars, and saturated fat. It is likely that those foods
... Read more5 Common Pitfalls When Importing Natural Health Products
As a license holder for Natural Health Products (NHPs), it is your responsibility to be aware of and educated on all requirements for Canada. This blog will aim to highlight and summarize the common pitfalls of a company that manufactures NHPs outside of and imports them into Canada. Working with an importer such as Quality
... Read moreInsider scoop on Selling, Importing & Registering your VHPs in Canada

Introduction: The market for animal and pet products is growing at a rapid pace. With that, there is also a constant need for compliance, with many Veterinary Health Products being rushed to market. This blog will aim to steer you toward compliance with your Veterinary Health Products (VHPs) or Animal Supplements in Canada. We will
... Read moreLearn how to differentiate Dietary Supplement vs NHP Labeling
Introduction Dietary supplements in the USA are very similar to natural health products (NHP regulations) in Canada and many products can be classified as both. But not all dietary supplements are natural health products and vice-versa. There are some distinct classification factors for each. Generally, dietary supplements are labeled comparably to food products in the
... Read moreFDA Qualified Health Claim for Magnesium 2022
The U.S. Food and Drug Administration (FDA) has communicated via Letter of Enforcement Discretion that it will not oppose or object to some qualified health claims regarding magnesium and the reduced risk of high blood pressure (hypertension), so long that the claims are worded properly to avoid misleading the consumer. The claims are essentially accompanied
... Read more11 Ways MoCRA will enhance FDA Cosmetics Regulations in 2023

Introduction: The Consolidated Appropriations Act, recently passed by the US Congress, incorporates revisions to US cosmetics laws. The Modernization of Cosmetics Regulation Act of 2022 (MoCRA), which has been in the works for years, is the biggest change to cosmetics legislation in recent U.S. history. The U.S. Food and Drug Administration (FDA) can now impose
... Read moreUpdate on Performance Standards for NHP Applications

Summary of Health Canada Licensing and Update Reports Product Licensing A combined 1390/5045 (28%) of NHP product license application submissions were refused from April to September 2021. Class I – 90% of product applications are meeting the 60-day performance timeline. The refusal rate for these is 14%. Class II – 90% of applications are meeting
... Read moreGreens and Proteins – Food or NHP?

The classification of Foods and Natural Health Products (NHPs) can be considered difficult, grey, and convoluted. Sometimes even comes down to preference of classification and labelling verbiage for certain ingredients. Foods are governed by the Food and Drug Regulations, however, NHPs fall under the Natural Health Product Regulations. Foods are thought to be consumed by
... Read moreHow Pet Food VHP Labels Differ from Human Food Labels in 2022
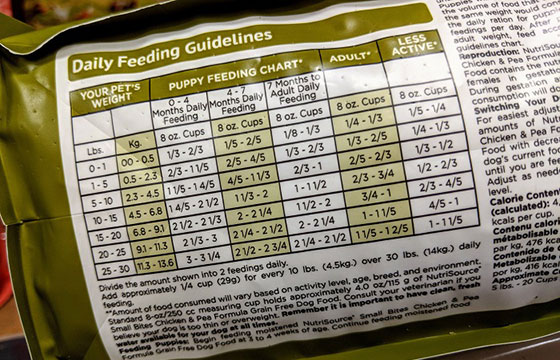
Pet Foods and Human Food Labels are designed to look similar yet distinct. There are some similar label requirements and some that will appear different as they are product specific. Consumer packaging rules in Canada aim to ensure we know what we are buying at time of purchase, so that the consumer can make educated
... Read moreBenefits of Routine Inspection of your Licensed Food Facility

Inspection of your food facility will help identify and fix any quality or processing deviations found regarding food safety. Review of the processes and procedures will lead to higher quality products that are safe for Canadians. Internal/self-inspection is important in day-to-day operation to ensure all processes are flowing smoothly, however third-party inspections are important to
... Read moreVitamin D Limits for NHPs are now Higher

The “Sunshine” vitamin, Vitamin D, Vitamin D3 or Vitamin D2 as it is more commonly known. The term “sunshine” was coined by the sunlight’s ability to provide levels of Vitamin D based on the level of exposure a person has. The most widely used form of Vitamin D is Cholecalciferol, which is predominately sourced from
... Read moreSummary of Guidelines for Advertising Health Products

Advertising is something almost all people experience and view daily. Yet, it can be challenging to navigate this space as it can be unclear to identify the truthful claims from the misleading claims. Fortunately, in Canada, Health Canada is the regulatory authority for consumer advertising, more specifically, health products. How does Health Canada regulate marketing?
... Read moreCFIA & Health Canada Policy on Food Labelling

Health Canada and the CFIA (Canadian Food Inspection Agency) are teaming up to plan advancements and improvements for the timing of food labelling changes in Canada. This is in accordance with the most recent 2019 Agri-food and aquaculture sector regulatory review roadmap. Changes of this nature may be required based on aligning with Canadian and
... Read moreWhat are the components of an NHP Label in 2023?
The product label is something we all look at in our daily lives, but not everyone is well equipped with the tools to read them efficiently. It can be challenging to try and navigate this space, especially for Natural Health Products (NHP), which can have multiple ingredients. First, you may be wondering what exactly is
... Read moreSupplemented Foods Proposed Regulatory Framework

Health Canada has been working on creating/revising the regulations with regards to Supplemented Foods for some time now. A recent Health Canada webinar indicated new proposals/consultation could be on the horizon. Health Canada anticipates publishing a new draft regarding the Supplemented Foods Regulations shortly (June 2021) in Canada Gazette Part I. Until now, supplemented foods
... Read moreProposed Food Safety Guidance for Novel Food Regulations Focused on Plant Breeding

CFIA and Health Canada have been working on new Food Safety guidance documents pertaining to Novel foods focused on plant breeding. Two new guidance documents are available to aid in your company’s plant breeding compliance, these are: Guidance on foods derived from products of plant breeding. Guidance on the pre-market assessment of food products derived
... Read morePerformance Standards for NHP Applications

The coronavirus-19 (COVID-19) pandemic has had a long-lasting effect on many of the processes done in business and everyday life. One of the resulting main effects is delays in application processing times. From simple applications such as passports to larger-scale applications such as NHP licensing. Prior to the pandemic, Natural and Non-Prescription Health Products Directorate
... Read moreComing into Force: Human Milk Fortifiers Regulations

After consultation with stakeholders and based on a noticeable lack of current regulations and need for availability and innovation for these types of products, Health Canada has released new Directives/Guidance Documents and Publications in Canada Gazette part II. These articles pave the way for new regulatory requirements and now list Human Milk Fortifiers (HMFs) within
... Read moreVeterinary Health Products to be allowed in Livestock Feed
Health Canada and the Canadian Food Inspection Agency have joined forces to evaluate the incorporation of Veterinary Health Products (VHPs) into Livestock Feeds (Food). This Project originated from an expressed interest by the industry to allow active VHP ingredients into Livestock feeds. Since the successful roll-out of the more recent VHP Notification Program, many Health
... Read moreInterim Order Respecting Clinical Trials Relating to COVID-19

On May 23, 2020, the Minister of Health of Canada approved an Interim Order (IO) Respecting Clinical Trials for Medical Devices and Drugs Relating to COVID-19. The IO aims to facilitate clinical trials for potential COVID-19 drugs and medical devices, while upholding strong patient safety requirements and validity of trial data. Under the IO, Health
... Read moreHealth Canada expedites access to COVID-19 diagnostic laboratory test kits and other medical devices
Recently in Canada, the Minister of Health has signed an Interim Order to allow expedited access to COVID-19-related medical devices for use by healthcare providers, including diagnostic test kits. This Interim Order will help ensure quicker and more flexible approval of the importation and sale of medical devices that are necessary for Canada’s response to
... Read more