




When thinking about NDI Classification have you ever heard about the New Dietary Ingredient Notification in Ingredient Compliance? In today’s rapidly evolving world, businesses constantly seek innovative ways to gain a competitive edge. One such method is harnessing the power of NDI classification. NDI, or New Dietary Ingredient, refers to any ingredient not marketed in the United States before October 15, 1994. Understanding the intricacies of NDI classification can be a game-changer for businesses in the dietary supplement industry. It ensures compliance with FDA regulations and opens up endless possibilities for product development and market expansion.
In this comprehensive guide, we will dive deep into the world of NDI classification, exploring its importance, the regulatory landscape surrounding it, and how businesses can navigate the complex process of submitting NDI notifications. Whether you are a small startup or an established player in the industry, this guide will equip you with the knowledge and tools to unlock the full potential of NDI classification and propel your business to new heights. Get ready to revolutionize your approach to dietary supplements and witness the transformative impact of NDI classification.
Why is NDI Classification important for businesses?
NDI classification is a crucial aspect of the dietary supplement industry that businesses cannot afford to overlook. The classification process determines whether an ingredient is considered “new.” It requires notification to the FDA before it can be legally marketed. This step is vital for businesses for several reasons. Firstly, it ensures compliance with FDA regulations, reducing the risk of penalties, recalls, or legal action. Secondly, the NDI classification provides a level playing field for all businesses, preventing unfair competition and protecting consumer safety. Following the NDI classification process, companies demonstrate their commitment to product quality and regulatory compliance, enhancing their reputation and building consumer trust.
Implementing NDI classification also offers businesses a competitive advantage. By thoroughly understanding the regulatory requirements and submitting NDI notifications, companies can introduce innovative ingredients and formulations to the market. This allows them to differentiate their products and tap into new consumer trends, ultimately driving sales and market share. Additionally, NDI classification opens opportunities for partnerships and collaborations with research institutions and other industry players. By showcasing their commitment to safety and compliance, businesses can attract investment and forge strategic alliances that amplify their growth potential.
In summary, NDI classification is a regulatory obligation, and a strategic tool businesses can leverage to enhance their reputation, drive innovation, and gain a competitive edge in the dietary supplement industry.
What does the NDI Classification process consist of?

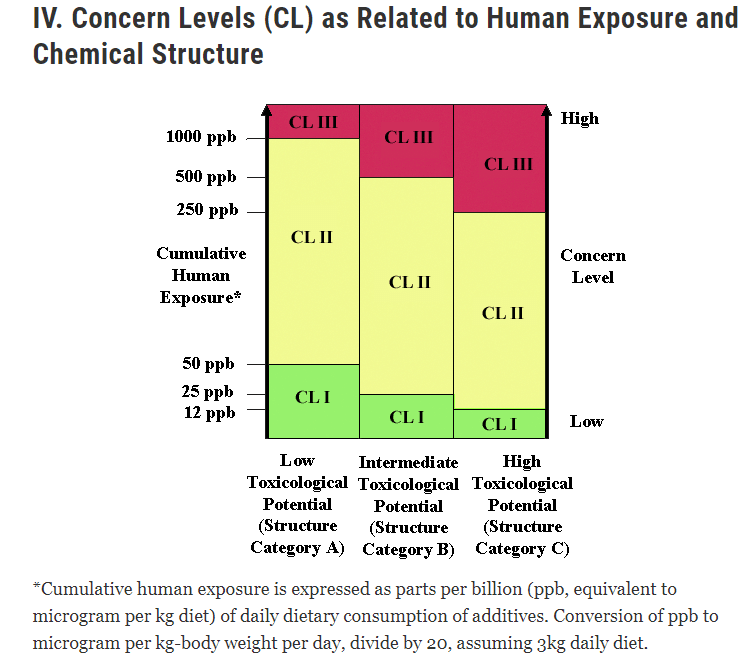
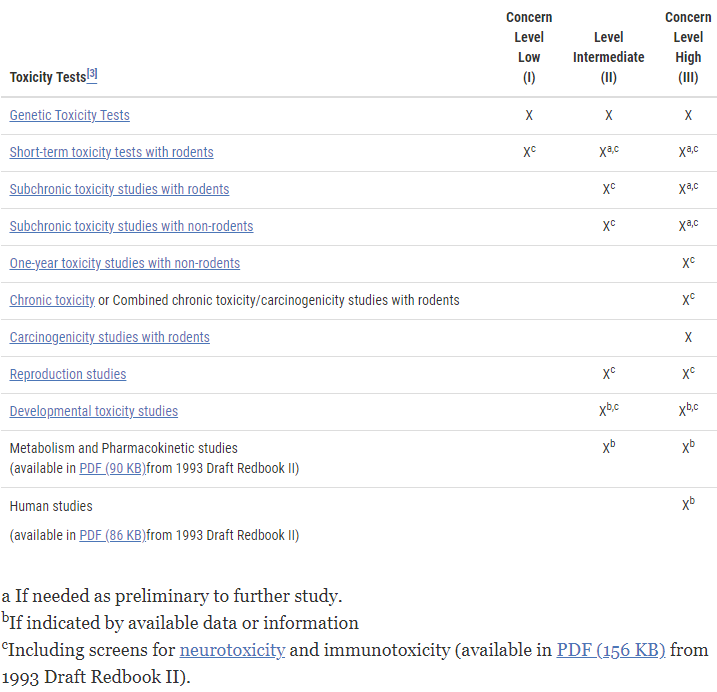
The NDI classification process involves several steps that businesses must carefully navigate to ensure compliance and successful market entry. The first step is to determine whether an ingredient qualifies as an NDI. As per FDA guidelines, an NDI is an ingredient not marketed in the United States before October 15, 1994. To establish this, businesses must thoroughly review historical data, including scientific literature, patents, and industry reports. This process helps companies to determine if their ingredient meets the criteria for NDI classification.
Once an ingredient is identified as an NDI, the next step is to prepare an NDI notification to the FDA. This notification must include detailed information about the ingredient, its safety profile, and the intended conditions of use. The message should address any potential concerns or risks associated with the ingredient and provide supporting scientific evidence to demonstrate its safety. The FDA evaluates the notification and determines if the NDI meets the safety standards required for market entry. Businesses must ensure that their notifications are comprehensive, scientifically sound, and meet all the regulatory requirements to increase the chances of approval.
Navigating the NDI classification process can be complex and time-consuming. It requires a deep understanding of FDA regulations, scientific expertise, and meticulous documentation. However, businesses can streamline the process by seeking guidance from experts in NDI classification or leveraging specialized software and tools that automate data collection, analysis, and notification preparation. By investing in the right resources and expertise, businesses can ensure a smooth and efficient NDI classification process, reducing the risk of delays or rejections and accelerating time to market.
What are the benefits of implementing NDI classification?
Implementing NDI classification offers businesses a myriad of benefits that go beyond regulatory compliance. One of the key advantages is the ability to introduce innovative ingredients and formulations to the market. NDI notifications allow businesses to showcase their scientific research, safety data, and unique selling points. By differentiating their products with NDI ingredients, companies can attract consumers looking for novel and effective dietary supplements. Differentiation allows businesses also to expand their product portfolios, tap into new market segments, and drive growth.
Another benefit of NDI classification is improved consumer trust and confidence. Consumers are becoming more discerning in their purchasing decisions with increasing product safety and quality scrutiny. Following the NDI classification process, businesses are committed to transparency, scientific rigor, and consumer safety. This builds trust with consumers, giving them peace of mind that the products they purchase are backed by rigorous scientific evaluation and meet the highest quality standards.
Furthermore, NDI classification can help businesses mitigate legal and regulatory risks. By complying with FDA regulations and submitting NDI notifications, companies reduce the likelihood of penalties, recalls, or legal action. This protects the business’s financial interests and safeguards its reputation and brand equity. Additionally, NDI classification fosters a culture of compliance within the organization, ensuring that all products meet regulatory requirements and adhere to the highest quality and safety standards.
In summary, implementing the NDI classification offers businesses a competitive advantage, enhanced consumer trust, and protection from legal and regulatory risks. By embracing the NDI classification, companies can unlock new market opportunities, drive growth, and establish themselves as leaders in the dietary supplement industry.
Tips for effective NDI classification
Navigating the NDI classification process can be daunting. Still, businesses can ensure a successful outcome with the right approach and resources. Here are some tips to effectively implement NDI classification:

1. Start with thorough research: Conduct comprehensive research to identify potential NDI ingredients before embarking on the NDI classification process. Review scientific literature, patents, and industry reports to gather evidence and understand the safety profiles of these ingredients.
2. Consult experts: Seek guidance from experts in NDI classification who can provide insights, review your scientific data, and help prepare comprehensive notifications. Their expertise can streamline the process and increase the chances of success.
3. Follow FDA guidelines: Familiarize yourself with FDA guidelines on NDI classification and ensure that your notifications meet all the regulatory requirements. Pay attention to the specific data and information that must be included in the message, such as safety data, intended conditions of use, and supporting scientific evidence.
4. Invest in resources: Consider investing in specialized software and tools that facilitate data collection, analysis, and notification preparation. These resources can streamline the process, reduce manual errors, and improve efficiency.
5. Maintain documentation: Keep detailed records of your NDI classification process, including supporting scientific data, safety evaluations, and communication with experts or consultants. This documentation is evidence of compliance and can be invaluable during FDA inspections or audits.
By following these tips, businesses can navigate the NDI classification process effectively, increase their chances of success, and unlock the transformative power of NDI classification.
Outsourcing NDI classification services
Outsourcing NDI classification services is a viable option for businesses needing more internal expertise or resources to navigate the NDI classification process effectively. Outsourcing allows companies to leverage the expertise of specialized consultants like Quality Smart Solutions or firms specializing in NDI classification. These experts can navigate regulatory requirements, conduct scientific evaluations, and prepare comprehensive notifications on behalf of the business.
When outsourcing NDI classification services, it is crucial to choose a reputable and experienced partner with decades of experience like us. We have a proven track record of successful NDI classifications, industry expertise, and a deep understanding of FDA regulations. We have experience with similar ingredients and can provide comprehensive support. Outsourcing NDI classification services can provide businesses peace of mind, ensuring compliance with FDA regulations and freeing up internal resources for core business activities.
Conclusion: Harnessing the power of NDI Classification for business success
In conclusion, NDI classification is a powerful tool that businesses in the dietary supplement industry can harness to gain a competitive edge, drive innovation, and achieve business success. By understanding the importance of NDI classification, navigating the complex process, and implementing effective strategies, businesses can unlock new market opportunities, enhance consumer trust, and ensure compliance with FDA regulations.
This guide has provided a comprehensive overview of NDI classification, covering its importance, the regulatory landscape, and the process of submitting NDI notifications. We explored the benefits of implementing NDI classification, showcased real-world case studies, and provided tips for effective implementation. Additionally, we highlighted tools and software options that streamline the NDI classification process and discussed training, certification, and outsourcing options.
Now armed with this knowledge, it’s time for businesses to embrace NDI classification, revolutionize their approach to dietary supplements, and unlock the full potential of this transformative tool. By integrating NDI classification into their business strategies, businesses can position themselves as leaders in the industry, drive growth, and meet the evolving needs of consumers. The power of NDI classification awaits – it’s time to unlock it and propel your business to new heights.
We hope this post helped you understand food additives and preservatives a little better. There are many different kinds, and they can have a big impact on your health and the environment. If you want to learn even more, reach out to us today!





 In marketing, the power of persuasive
In marketing, the power of persuasive 

 In a world where the pharmaceutical industry is constantly evolving and new drugs are being introduced to the market every day, the role of
In a world where the pharmaceutical industry is constantly evolving and new drugs are being introduced to the market every day, the role of 
 Are you a food, beverage, drug, or cosmetics business owner?
Are you a food, beverage, drug, or cosmetics business owner?








 Are you a company in the healthcare or pharmaceutical industry looking to bring a new product to market? If so, you’re probably aware of the complex and ever-changing regulatory landscape governed by the Food and Drug Administration (FDA). Navigating the FDA’s requirements is a challenging task, and one aspect that often catches businesses off guard is the associated fees and costs. Budgeting and planning for these regulatory expenses can be daunting, especially for startups and small businesses.
Are you a company in the healthcare or pharmaceutical industry looking to bring a new product to market? If so, you’re probably aware of the complex and ever-changing regulatory landscape governed by the Food and Drug Administration (FDA). Navigating the FDA’s requirements is a challenging task, and one aspect that often catches businesses off guard is the associated fees and costs. Budgeting and planning for these regulatory expenses can be daunting, especially for startups and small businesses.














 Introduction:
Introduction:


 As a business owner or operator, you may know the importance of ensuring that your products and services meet regulatory requirements. However, regarding the Canadian Consumer Product Safety Act (CCPSA), compliance is not just substantial – it’s crucial. The CCPSA is a federal law that governs the safety of consumer products sold in Canada, and it applies to a wide range of products, from toys and household appliances to food and cosmetics.
As a business owner or operator, you may know the importance of ensuring that your products and services meet regulatory requirements. However, regarding the Canadian Consumer Product Safety Act (CCPSA), compliance is not just substantial – it’s crucial. The CCPSA is a federal law that governs the safety of consumer products sold in Canada, and it applies to a wide range of products, from toys and household appliances to food and cosmetics.







