The Licensing and Non-paired Health Products Directorate (LNHPD) is a federal government organization that oversees the regulation of natural health products in Canada. It works to promote quality, safety, and efficacy in natural health products while providing Canadians with access to safe, effective, and high-quality products.
If you’re in the business of labeling natural health products in Canada, you’ll need to know about the NHP Database. This centralized platform provides important information for consumers and regulators alike, so it’s important to make sure your product is included. But how do you submit your product to the database? Here’s everything you need to know.
What is the LNHPD (NHP Database) and its Role in Canada’s Health System?
The Licensing and Non-paired Health Products Directorate (LNHPD) is a federal government organization whose fundamental role is to regulate natural health products in Canada. It works to ensure that all natural health products sold in Canada are of high quality, safe, and effective while providing Canadians access to safe and reliable products. LNHPD also monitors the manufacturing, importing, labeling, and advertising of natural health products so that companies comply with the applicable laws. Furthermore, it supports research initiatives in natural health products to facilitate a better understanding of their effects on human health.
The NHP Database is a centralized platform that contains information about natural health products available in Canada. It was created to provide consumers and regulators with easy access to important safety and efficacy information on these products. By submitting to get your NHP product license you get your product in the database, and you are ensuring that it is included by Health Canada in this important resource for consumers and regulators alike.
Who can submit products to the LNHPD (NHP Database)?
Anyone can submit a natural health product to the NHP Database in Canada, regardless of whether they are the manufacturer or distributor. However, the submitter must ensure that the product meets all regulatory requirements set out by Health Canada. This includes providing accurate and complete information about the product’s ingredients, dosage form, recommended use, and warnings or contraindications. It’s important to note that all submissions must be made online using Health Canada’s Natural and Non-prescription Health Products Directorate (NNHPD) website.
What are the requirements for submitting a product to the NHP Database (LNHPD)?
If you want to submit your product to the NHP Database in Canada, certain requirements must be met. First and foremost, you need to ensure that your natural health product complies with all regulatory requirements set by Health Canada. This includes providing detailed information about the product such as its ingredients, dosage form, recommended use, and warnings or contraindications. Keep in mind that all submissions must be made online using Health Canada’s Natural and Non-prescription Health Products Directorate (NNHPD) website. Additionally, you should also provide evidence to support the safety and effectiveness of your product including supporting data from clinical trials or published studies. By meeting these requirements, you can assist the submission process and get your product approved for sale.
How to fill out and submit a product to the NHP Database (LNHPD)?
If you want to submit your natural health product to the NHP Database, the first step is to ensure that it complies with all regulatory requirements set by Health Canada. Next, you can visit the Natural and Non-prescription Health Products Directorate (NNHPD) website to fill out an electronic application form. Before starting your application, make sure that you have all the necessary details about your product such as its ingredients, dosage form, recommended use, and warnings or contraindications. It’s also important to provide supporting evidence to support the safety and effectiveness of your product.
This can include data from clinical trials or published studies. Once your application is complete and submitted, it will be reviewed by the NNHPD. You may need to provide additional information or respond to questions during this process. If your application is approved, your product will be added to the NHP Database and will be authorized for sale in Canada.
Tips for a successful product submission and approval process:
Submitting your natural health product to the NHP Database can be a lengthy process, but there are steps you can take to increase your chances of a successful submission and approval. Firstly, ensure that your product meets all regulatory requirements set by Health Canada. Next, make sure you have all the necessary details about your product ready before starting your application. This includes its ingredients, dosage form, recommended use, and warnings or contraindications. Providing supporting evidence for the safety and effectiveness of your product is also crucial. This can include data from clinical trials or published studies. If during the review process, additional information or questions arise from the NNHPD, make sure to respond promptly with accurate information. By following these steps diligently you will increase the likelihood of having your product approved and added to the NHP database for sale in Canada.
What Standards Must Natural Health Products Meet Before Licensing?
Before a natural health product is licensed and allowed to be sold, it must meet the requirements set out in the Natural Health Products Regulations. These include manufacturing quality standards, such as Good Manufacturing Practices (GMP); labeling standards, such as accurate and complete information about the product, including all ingredients and potential risks associated with taking it; and marketing standards, such as advertising that is accurate and not deceptive. In addition, natural health products must also contain standardized labels of their ingredients, so consumers can be confident they are getting what they expect when using a product.
How Does the LNHPD Ensure Quality of Natural Health Products?
The LNHPD is responsible for monitoring and regulating natural health products, and ensuring they meet the requirements set out in the Natural Health Products Regulations. They evaluate each product submitted to them, which includes analyzing its ingredients and its safety. They also inspect manufacturing sites to ensure that they are following GMP practices and have protocols in place to ensure the quality of their finished products. In addition, they review advertising to make sure it is factual and not misleading, with no false claims or exaggerations promoting their products.
What Information Does a Company Need to Provide for Product Licensure in the NHP Database?
To obtain a product license from the LNHPD, a company will need to provide comprehensive information about its product, its ingredients, and how it is manufactured. This includes evidence that each ingredient has been evaluated for safety and efficacy in terms of clinical trials or evidence from traditional medical practice. Once these documents have been submitted to the LNHPD, they will conduct a detailed review process before granting a product license.
Is There an Appeal Process if an Application for Licensure is Refused?
Yes, there is an appeals process if a company’s application for licensure is refused. The appeals process is handled by the Product Licensing Appeal Committee (PLAAC). This committee has the power to review the decision and offer a set of recommendations to the LNHPD regarding the application. Companies that feel their license was wrongly denied can file an appeal with PLAAC and should submit any additional documents or evidence to present their case.
How can I ensure successful product submission and approval process for the LNHPD?
Submitting your natural health product to the NHP Database can be a lengthy process, but there are steps you can take to increase your chances of a successful submission and approval. Firstly, ensure that your product meets all regulatory requirements set by Health Canada. Next, make sure you have all the necessary details about your product ready before starting your application. This includes its ingredients, dosage form, recommended use, and warnings or contraindications. Providing supporting evidence, the safety and effectiveness of your product are also crucial. This can include data from clinical trials or published studies. If during the review process, additional information or questions arise from the NNHPD, make sure to respond promptly with accurate information. By following these steps diligently you will increase the likelihood of having your product approved and added to the NHP database for sale in Canada.
What are NHPD monographs?
NHPD monographs are documents created by the Natural Health Products Directorate (NHPD) in Canada that outline the safety, efficacy, and quality requirements for natural health products. These monographs guide manufacturers and distributors of natural health products, helping them ensure that their products meet the necessary standards for safety and effectiveness. Understanding and following these monographs is crucial for businesses in the natural health product industry to comply with regulations and maintain consumer trust. These documents provide information on the safety, efficacy, and quality of natural health products.
How do NHPD monographs impact the natural health product industry?
NHPD monographs have a significant impact on the natural health product industry. They provide clear guidelines for manufacturers and distributors to follow, ensuring that their products are safe and effective for consumers. Failure to comply with these monographs can result in legal consequences, including fines and product recalls. Additionally, following NHPD monographs can help businesses build consumer trust and establish a positive reputation in the industry.
What information is included in NHPD monographs?
NHPD monographs include detailed information about natural health products, including their ingredients, recommended uses, and potential side effects. They also outline the specific requirements that manufacturers and distributors must meet to ensure the safety and efficacy of their products. This information is based on scientific research and is regularly updated to reflect new findings and developments in the industry. By following NHPD monographs, businesses can ensure that their products are of the highest quality and meet the needs of their customers.
How can businesses use NHPD monographs to their advantage?
By following NHPD monographs, businesses can ensure that their products are safe, effective, and meet the standards set by Health Canada. This can help build trust with customers and improve the reputation of the business. Additionally, by staying up-to-date with the latest research and developments in the industry, businesses can stay ahead of the competition and offer innovative products that meet the changing needs of consumers. Overall, understanding and following NHPD monographs is essential for any business in the natural health product industry.
What are the NHPD regulations and guidelines?
The Natural Health Products Regulations (NHPR) are the guidelines that govern the sale, distribution, and importation of natural health products in Canada. These regulations require that all natural health products be licensed by the Natural and Non-prescription Health Products Directorate (NNHPD) before they can be sold in Canada. The NNHPD also guides the safety, efficacy, and quality of natural health products through the use of monographs. These monographs provide detailed information on the ingredients, recommended uses, and potential side effects of natural health products.
How to read and interpret NHPD monographs?
Reading and interpreting NHPD monographs can be overwhelming, but it’s important to understand them if you’re involved in the natural health products industry. Monographs typically include information on the product’s active ingredients, recommended uses, dosage, and potential side effects. It’s important to pay attention to any warnings or precautions listed in the monograph, as well as any contraindications or interactions with other medications. If you’re unsure about how to interpret a monograph, consult with a healthcare professional or regulatory expert.
What are some NHPD monograph key ingredients and their benefits?
NHPD monographs typically list the active ingredients in natural health products and their potential benefits. Some common ingredients include echinacea, which is believed to boost the immune system and reduce the duration of colds and flu; St. John’s Wort, which is used to treat mild to moderate depression; and ginger, which is used to relieve nausea and vomiting. It’s important to note that while these ingredients may have potential benefits, they can also have side effects and interactions with other medications, so it’s important to use them under the guidance of a healthcare professional.
Tips for using NHPD monographs in product development:
When using NHPD monographs in product development, it’s important to keep in mind that they are not a substitute for clinical trials or other forms of scientific research. Monographs provide a general overview of the potential benefits and risks of natural health products, but they do not provide specific information about the safety and efficacy of individual products. It’s also important to ensure that any ingredients used in product development are sourced from reputable suppliers and meet all regulatory requirements. Finally, it’s important to consult with a healthcare professional or regulatory expert to ensure that your product complies with all relevant regulations and guidelines.
What are the potential consequences of not following NHPD monographs?
Not following NHPD monographs can have serious consequences for businesses in the natural health product industry. Health Canada may take enforcement action, which can include product seizure, fines, and even criminal charges. Additionally, not following NHPD monographs can damage the reputation of the business and lead to a loss of customer trust. Businesses need to stay informed and compliant with NHPD monographs to ensure the safety and effectiveness of their products and the success of their business.
Get In Touch with Experts for FREE Consultation
By familiarizing yourself with the regulatory landscape, you can be confident that you are using natural health products that meet the highest standards of safety and quality.
For 6 important things to know about your NPN and the NHPID click here:


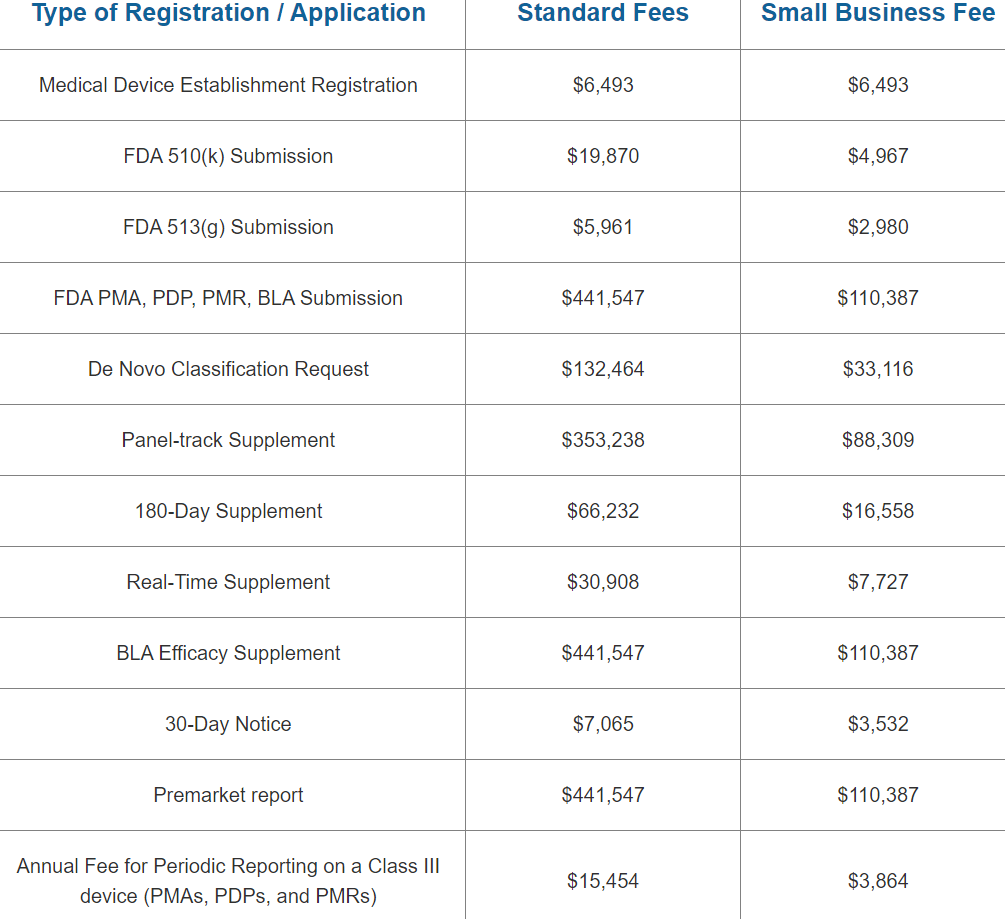
 As a business owner, keeping up with regulatory requirements is a top priority. One such requirement is the FDA’s renewal registration. The process of registering your facility with the FDA can be confusing, but it’s essential to comply in order to avoid penalties or even shutting down your operation.
As a business owner, keeping up with regulatory requirements is a top priority. One such requirement is the FDA’s renewal registration. The process of registering your facility with the FDA can be confusing, but it’s essential to comply in order to avoid penalties or even shutting down your operation. 





 Introduction:
Introduction: