
HACCP Implementation: A Practical Guide for Food Businesses

HACCP, which stands for Hazard Analysis and Critical Control Points, is the cornerstone of food safety plans in the food industry. It is a concept designed to identify and prevent or eliminate any potential hazards that may put consumers at risk of food-borne illnesses. This guide will take an in-depth look into what HACCP is
... Read more5 FDA Warning Letters: Food Manufacturers of CBD Food Products

Introduction On November 21, 2022, the Food and Drug Administration (FDA) Center for Food Safety and Applied Nutrition issued warning letters to food manufacturers who were selling products containing cannabidiol (CBD). These letters were issued in response to an online search for companies selling foods or beverages containing CBD as a continued FDA CBD enforcement
... Read moreSafe Quality Food Certification SQF Food Safety Program

Food is one of the most important parts of our lives. We eat it to stay alive, and we eat it to enjoy life. Unfortunately, foodborne illnesses are a major issue in Canada, taking a toll on our health and economy. One way to reduce the risk of foodborne illnesses is to certify your food
... Read moreFDA UDI Labeling Requirements for FDA Class 1 Medical Device in 2023

Introduction: FDA regulations for Class 1 medical devices can be daunting and confusing to navigate. However, with this easier-to-understand overview, you’ll gain insight into the prerequisites for device categories, UDI labeling requirements, and more. With the proliferation of medical devices worldwide, it is important to stay on top of applicable UDI labeling requirements. This
... Read moreUnderstanding MDALL/MDEL & How It Transforms Your Business (MDL, MDALL, MDEL Listing)
Navigating medical device regulations set by Health Canada can be challenging. However, knowing the correct classification of your medical device is essential. Proper classification ensures your device complies with regulatory requirements. MDALL provides a strong solution for businesses looking to improve operations. It supports automated workflows, better customer service, and higher productivity. Using MDALL can
... Read moreMedical Device License (MDL MDEL SaMD) Under the Microscope
The Medical Device License is a legal document required to manufacture, sell, and distribute medical devices. The Medical Device Licence (MDL) and the Medical Device Establishment Licence (MDEL) are the licenses issued by Health Canada, the Health Agency that regulates the medical devices sold in the Canadian market. These licences are referred to as “medical
... Read moreCFIA Enforcement of Nutrition Facts Table & CFIA Labeling Requirements
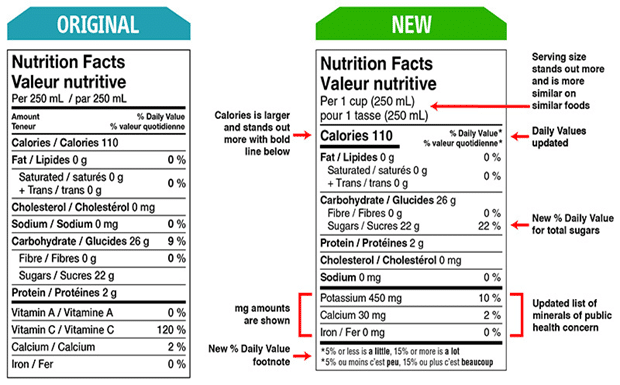
The Canadian Food Inspection Agency (CFIA) protects Canadians’ health and safety by ensuring food that reaches customers is safe, nutritious, and labeled. The CFIA’s responsibilities also include enforcing federal food safety and nutrition regulations. As of July 1, 2016, the CFIA has begun to enforce the new Nutrition Facts Table (NFT) and food labeling regulations. All
... Read moreFSVP Compliance: Regulatory Assessment Record Keeping 2023

U.S.A RRA FDA Draft Guidance: In a recent U.S. Food and Drug Administration (FDA) draft guidance detailing its implementation of Remote Regulatory Assessments (RRAs). Questions and answers outlined the regulatory oversight during and after the COVID-19 pandemic. The draft, “Conducting Remote Regulatory Assessments: Questions and Answers,” outlines the FDA’s use of RRAs to continue
... Read moreHow the Cosmetic Notification Form Works in Canada

Top Canada Cosmetic Regulations Cosmetic Notification Form (CNF) Canada is one of the most popular tourist destinations in the world, and it’s no surprise that the country has some of the most stringent cosmetic regulations in the world. In this blog post, we’ll look at some of the top Canadian cosmetic regulations and what you
... Read moreFood Labelling & Health Canada Packaging Update for 2023

Health Canada Announcement: The Minister of Health has introduced new front-of-package nutrition labeling laws for prepackaged food products, and they will apply to packaged foods high in saturated fat, sugars, and salt. A front-of-package emblem will need to be shown on these prepackaged foods in order to comply with the new requirements. Food products
... Read moreFDA NAC Warning Letter: Targeting N-Acetyl Cysteine (NAC) Supplements

Background on FDA NAC Warning Letters: Navigating the FDA’s NAC system can be overwhelming. Understanding FDA warning letters is key to ensuring compliance, avoiding costly penalties and maintaining a successful business relationship with federal regulators. This guide will teach you how to understand FDA warning letters and provide guidance on how to comply. N-acetyl-L-cysteine (NAC
... Read moreInside Scoop FDA Medical Device User Fees (MDUFA) for 2023
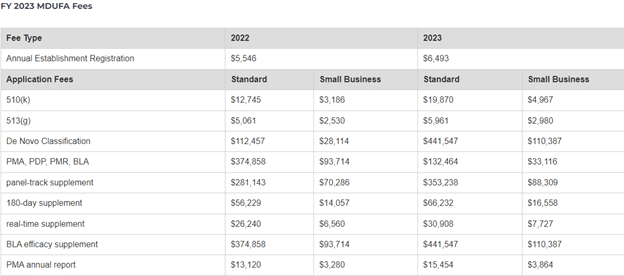
The FDA or U.S. Food and Drug Administration recently announced the Fiscal Year Medical Device User Fee (MDUFA) amendments. The fiscal year will begin on October 1st, 2022, and end on September 30th, 2023. All medical device facilities will have to pay these fees to remain FDA-compliant. In this blog, we’re going to elaborate on
... Read more