
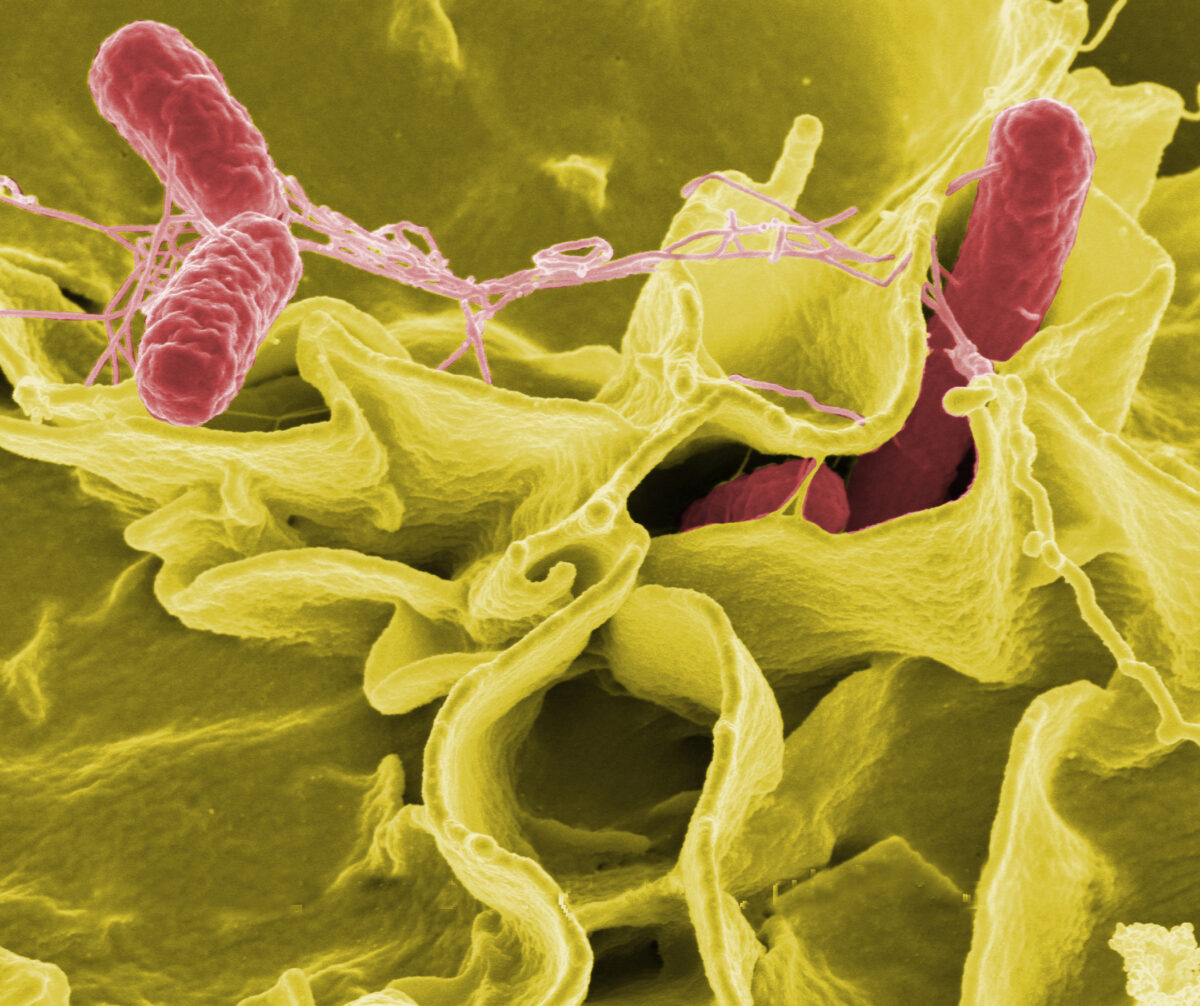
The Interagency Food Safety Analytics Collaboration (IFSAC), a joint initiative of the FDA, CDC, and USDA-FSIS, has recently released its 2022 Annual Report on Foodborne Illness Source Attribution Estimates. This comprehensive report sheds light on the primary sources of foodborne illnesses caused by Salmonella, Escherichia coli O157, and Listeria monocytogenes, offering critical insights for food safety stakeholders.
Foodborne Pathogens and Their Sources
The 2022 report highlights the leading food categories associated with these pathogens, providing actionable data to reduce foodborne illness outbreaks:
Salmonella
- Top Sources: Chicken, turkey, and seeded vegetables.
- Impact: One of the most prevalent causes of foodborne illnesses and hospitalizations, causing diarrhea, fever, and abdominal cramps. Can be life-threatening in young children, the elderly, and those with weakened immune systems.
- Key Considerations: Salmonella is often present in the intestines of animals, making cross-contamination a significant concern during food processing.
- By The Numbers/Annual Statistics: Salmonella causes an estimated 1.35 million illnesses, 26,500 hospitalizations, and 420 deaths in the United States every year.
- Prevention Strategies: Consumers should cook poultry thoroughly to 165°F (74°C), wash hands and surfaces after handling raw poultry, and thoroughly wash seeded vegetables. The industry should implement strict hygiene practices during processing and use appropriate antimicrobial treatments.
E. coli O157
- Top Sources: Leafy greens and beef.
- Impact: Causes severe diarrhea (often bloody), abdominal cramps, and vomiting. Can lead to kidney failure in young children.
- Key Considerations: Can survive in acidic environments and at low temperatures, making it challenging to eliminate.
- Prevention Strategies: Consumers should thoroughly wash leafy greens, cook beef to an internal temperature of 160°F (71°C), and avoid cross-contamination between raw meat and other foods. The industry should implement stringent hygiene practices during cultivation and processing of produce and use effective sanitation methods in beef processing plants.
- By The Numbers/Annual Statistics: E. coli O157 causes an estimated 63,153 illnesses, 2,138 hospitalizations, and 20 deaths in the United States each year.
Listeria monocytogenes
- Top Sources: Dairy products (especially soft cheeses), deli meats, and fruits.
- Impact: Causes fever, muscle aches, and sometimes gastrointestinal symptoms. Can lead to severe complications like meningitis and sepsis, especially in high-risk groups.
- Key Considerations: Listeria is unique because it can grow at refrigerator temperatures, making it a concern in ready-to-eat foods with longer shelf lives.
- Prevention Strategies: Consumers, especially high-risk individuals, should avoid soft cheeses, deli meats, and unpasteurized dairy products and thoroughly wash fruits before consumption. The industry should maintain strict temperature control during processing and storage and implement environmental monitoring programs to detect Listeria in processing facilities.
- By The Numbers/Annual Statistics: Listeria causes an estimated 1,600 illnesses, 1,500 hospitalizations, and 260 deaths in the United States annually.
How This Report Helps Improve Food Safety
The IFSAC report is a cornerstone resource for food industry professionals, public health officials, and policymakers. Its data enables:
- Enhanced Risk Management: Identifying high-risk foods to prioritize safety measures.
- Strengthened Compliance: Aligning practices with updated FDA and USDA regulations.
- Informed Decision-Making: Supporting data-driven strategies to mitigate contamination risks.
Empowering Food Safety Stakeholders
The report’s actionable insights regarding foodborne illness can be applied across the food supply chain:
- Manufacturers: Implement rigorous testing and preventive controls for high-risk products.
- Retailers: Educate staff and consumers on safe handling practices.
- Regulators: Focus inspections and resources on the most susceptible food categories.
A Call to Action for Safer Food Practices
IFSAC’s 2022 Annual Report emphasizes the importance of collaboration and proactive measures in combating foodborne illness. By addressing the identified sources, the food industry can play a pivotal role in reducing outbreaks and protecting public health.
To explore the full report and its findings, visit the FDA website.


 Understanding the FDA classification system for medical devices is key to getting your product to market safely and efficiently. Knowing how the
Understanding the FDA classification system for medical devices is key to getting your product to market safely and efficiently. Knowing how the 


 If you’re planning to bring veterinary health products (VHPs) into Canada, you’ve probably come across the terms VHP Canadian representative and VHP Importer of Record. Sounds technical, right? But don’t worry, it’s not as complicated as it seems! These roles may sound similar, but they have distinct responsibilities that are essential to your success in the Canadian market.
If you’re planning to bring veterinary health products (VHPs) into Canada, you’ve probably come across the terms VHP Canadian representative and VHP Importer of Record. Sounds technical, right? But don’t worry, it’s not as complicated as it seems! These roles may sound similar, but they have distinct responsibilities that are essential to your success in the Canadian market.
 Are you prepared for changes to Health Canada’s regulations on infant formulas and dietary products? The Interim Policy on Importation and Sale ends on December 31, 2024. To ensure compliance and long-term availability, you need to align your products with the Food and Drugs Regulations (FDR). Let’s explore what this means and how you can adapt.
Are you prepared for changes to Health Canada’s regulations on infant formulas and dietary products? The Interim Policy on Importation and Sale ends on December 31, 2024. To ensure compliance and long-term availability, you need to align your products with the Food and Drugs Regulations (FDR). Let’s explore what this means and how you can adapt.
 Selling natural health products (NHPs) on Amazon Canada is a lucrative opportunity to expand your business and reach millions of potential customers. However, navigating the process requires understanding and adhering to Canadian regulations, as well as Amazon’s specific policies for NHPs.
Selling natural health products (NHPs) on Amazon Canada is a lucrative opportunity to expand your business and reach millions of potential customers. However, navigating the process requires understanding and adhering to Canadian regulations, as well as Amazon’s specific policies for NHPs.
 Are you ready for the upcoming changes to Health Canada Fees in 2025? These fees, effective from April 2025, impact businesses dealing with drugs, medical devices, and cannabis. With updates spanning licensing, submissions, and compliance requirements, staying informed is key to avoiding disruptions.
Are you ready for the upcoming changes to Health Canada Fees in 2025? These fees, effective from April 2025, impact businesses dealing with drugs, medical devices, and cannabis. With updates spanning licensing, submissions, and compliance requirements, staying informed is key to avoiding disruptions.
